
Glomeruloesclerose segmentar e focal familiar: a associação com mutação no gene COL4A5, no interior da Amazônia
##plugins.themes.bootstrap3.article.sidebar##
##plugins.themes.bootstrap3.article.main##
Resumo
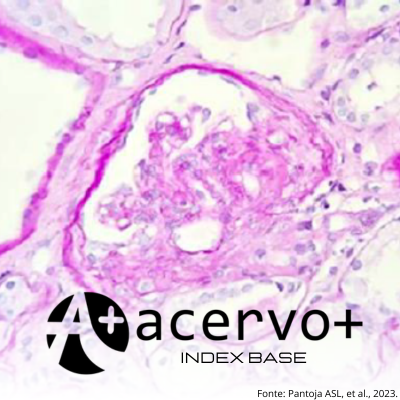
Objetivo: Relatar um caso de Glomeruloesclerose Segmentar e Focal (GESF) familiar no interior da Amazônia. Detalhamento de caso: O relato de um paciente (membro índice) com GESF diagnosticada por biópsia renal associada à Síndrome de Alport 1, além de dois irmãos portadores de Doença Renal Crônica de causa não esclarecida e outros cinco indivíduos a serem rastreados para síndromes glomerulares, incluindo quatro irmãos dessa mesma família e a sua mãe. O membro índice procurou atendimento como forma preventiva devido doença renal crônica de dois irmãos mais velhos e queixava-se de urina espumosa. Ao exame físico, mostrava-se hipertenso. Laboratorialmente, apresentava-se com proteinúria, hematúria, hipertrigliceridemia e insuficiência renal classe G3bA3. A biópsia renal, revelou achados compatíveis com Glomeruloesclerose Segmentar e Focal. No painel genético, observou-se, no gene COL4A5, a variante ChrX:108.603.036 G>T, associada à Síndrome de Alport 1. A mãe apresentava função renal alterada, proteinúria, hematúria e hipertensão arterial, e a irmã revelou discreta hipoalbuminemia e alteração do perfil lipídico. Considerações finais: A variante COL4A5 representa um fator de risco para o desenvolvimento de doença renal terminal precoce grave e parece não responder à corticoterapia convencional. O estudo permitiu o diagnóstico de um provável caso brando da doença no sexo feminino.
##plugins.themes.bootstrap3.article.details##
Copyright © | Todos os direitos reservados.
A revista detém os direitos autorais exclusivos de publicação deste artigo nos termos da lei 9610/98.
Reprodução parcial
É livre o uso de partes do texto, figuras e questionário do artigo, sendo obrigatória a citação dos autores e revista.
Reprodução total
É expressamente proibida, devendo ser autorizada pela revista.
Referências
2. CORIAT JA, et al. Nefropatia por C1q como diagnóstico diferencial de nefrite lúpica: um relato de caso. Revista Eletrônica Acervo Saúde, 2021; 13 (10): e8884.
3. CUNHA MFM, et al. Doenças renais hereditárias raras: um campo em evolução na Nefrologia. Brazilian Journal of Nephrology, 2020; 42: 219-230.
4. ELGALY SSJ, et al. Glomeruloesclerose segmentar e focal em associação a infecção pela Covid-19. Revista Eletrônica Acervo Saúde, 2022; 15 (3): e9935.
5. GARCIA J, et al. Glomeruloesclerose focal e segmentar: avanços no diagnóstico e consenso no tratamento. CuidArte, Enferm, 2020; 14(2): 265-269.
6. HUSSAIN HMJ, et al. Mutation in XPO5 causes adult-onset autosomal dominant familial focal segmental glomerulosclerosis. Human genomics, 2022; 16(1): 57.
7. JUNQUEIRA V, et al. Síndrome nefrótica após picada de inseto: relato de caso. Brazilian Journal of Nephrology, 2020; 42: 498-501.
8. KASHTAN CE. Alport syndrome: achieving early diagnosis and treatment. American Journal of Kidney Diseases, 2021; 77 (2): 272-279.
9. KDIGO. KDIGO 2021 Clinical Practice Guideline for the Management of Glomerular Diseases. Disponível em: https://kdigo.org/wp-content/uploads/2017/02/KDIGO-Glomerular-Diseases-Guideline-2021-English.pdf. Acessado em: 20 de maio de 2023.
10. LEPORI N, et al. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clinical kidney journal, 2018; 11 (2): 179-190.
11. LIMA HN, et al. Distribuição das nefropatias em Joinville, Santa Catarina: análise de um banco de dados de biópsia renal entre 2008 a 2019. Brazilian Journal of Nephrology, 2022; 44: 358-367.
12. NOZU K, et al. A review of clinical characteristics and genetic backgrounds in Alport syndrome. Clinical and experimental nephrology, 2019; 23: 158-168.
13. NOZU K, et al. Genetic background, recent advances in molecular biology, and development of novel therapy in Alport syndrome. Kidney Research and Clinical Practice, 2020; 39(4): 402.
14. PLEVOVÁ P, et al. Familial hematuria: a review. Medicina, 2017, 53(1): 1-10.
15. SAVIGE J, et al. Alport syndrome in women and girls. Clinical journal of the American Society of Nephrology: CJASN, 2016; 11(9): 1713.
16. SAVIGE J, et al. Genotype-phenotype correlations for pathogenic COL4A3–COL4A5 variants in X-linked, autosomal recessive, and autosomal dominant Alport syndrome. Frontiers in medicine, 2022; 9: 865034.
17. SAVIGE J, et al. Guidelines for genetic testing and management of Alport syndrome. The Clinical Journal of the American Society of Nephrology, 2022; 17(1): 143-154.
18. SIQUEIRA RTS, et al. Análise do perfil clínico e sociodemográfico dos pacientes pediátricos diagnosticados com glomerulonefrite difusa aguda em hospital no Sertão de Pernambuco, Brasil. Archives of Health Investigation, 2020; 9(5): 420-425.
19. WARADY BA, et al. Alport syndrome classification and management. Kidney medicine, 2020; 2 (5): 639-649.
20. WATSON S, et al. Síndrome de Alport. In: StatPearls [Internet]. Ilha do Tesouro (FL): StatPearls Publishing; 2023. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK470419/. Acessado em: 18 de junho de 2023.